

Discover
Visualize rapid, validated insights through real-world data.

An external control arm (ECA) study is a cutting-edge pharmacoepidemiologic design that has the potential to transform the way that new drugs get evaluated and approved, especially in rare indications. However, this term is often discussed without much knowledge about what an ECA is, or what creating and implementing a high quality ECA involves. In order to be robust and regulatory-grade, ECAs require access to fit-for-purpose data coupled with a high level of data fluency, methodological expertise, and advanced knowledge of a real-world evidence (RWE) analytics platform or program meant to support this type of work.
Implementing an ECA typically requires constructing a control arm—usually a group of patients taking the standard of care therapy to compare to those using a new or different therapy—for a single-arm trial out of real-world data (RWD), which is comprised of patients that could have been included in the clinical trial if the sponsor had enrolled one in the first place. ECAs are particularly important in rare disease areas, including oncology, where it can be difficult or unethical to enroll a placebo or control arm into the clinical trial.
Read on to learn more about constructing an ECA, and how RWE platforms provide the necessary guardrails to help design decision-quality studies.
Step 1: Choose a fit-for-purpose data source
The first step to building an ECA is identifying a complementary RWD source from which we can identify our control patients. This can involve consulting data strategists to comprehensively evaluate the potential datasets to make sure that they have both the richness and fields that are required for the study at hand. It’s also critical to ensure the dataset contains sufficient patients to power the study.
Step 2: Prepare the data for analysis
After selecting the data source(s), we need to build a common data model to bring the clinical trial data and RWD together into one, workable dataset that we can analyze. This is a particularly challenging process given that RWD is inherently different than clinical trial data. Patients in a clinical trial, especially in rare disease or oncology, are highly monitored on a regular schedule. On the other hand, patients not enrolled in clinical trials do not see their doctors as frequently, so they aren’t as closely monitored. Their clinicians also aren’t required to document patient encounters with the level of detail required in a trial, as clinicians are more focused on treating their patients than creating rich data sources to later be used in epidemiologic research.
To build the common data model, epidemiologists and data scientists collaborate to accurately and appropriately map all the key characteristics from the clinical trial dataset and the RWD together. These data can then be loaded into the RWE platform for analysis. To make this process more efficient, it’s helpful to use a platform that is data agnostic, such as the Aetion Evidence Platform® (AEP).
Step 3: Construct the control arm
Once we have the data in a workable format and accessible within the platform, we focus our efforts on building a two-armed patient population. In order to truly simulate the group of patients from the real world that could have been enrolled in the clinical trial, we must apply the inclusion and exclusion criteria of the trial to the real-world patients. If we’re using a fit-for-purpose data source, we should be able to apply the relevant criteria to the real-world patients while maintaining enough patients in our cohort to properly assess outcomes.
Step 4: Use propensity scores to ensure populations are comparable
Before we can look at outcomes, we need to be sure that the exposure arm (clinical trial patients) and control arm (real-world patients on standard of care therapies) are truly comparable. Comparability (or exchangeability) is crucial—when we look at results, we want to be able to say that any difference in outcomes must be due to the difference in treatment, because the patients are similar in all other ways. We do this with propensity score adjustment, either through matching or weighting, and the choice between the methods depends, in part, on the stakeholder audience.
In simple terms, propensity score matching involves looking at each patient in the clinical trial and trying to find them a “match” in the real world. The matched patient has similar characteristics to the clinical trial patient, such as their age, gender, smoking status, or comorbidities. Matched patients are then retained in the cohort for outcome analysis, while unmatched patients are dropped. This process can result in good balance and is easily understood by stakeholders. It does, however, risk losing clinical trial patients from the analysis if they cannot find a sufficient match in the real world. This can deter researchers from using the matching method, especially for studies in already small patient populations.
On the other hand, propensity score weighting involves up-weighting patients that are the most similar (and down-weighting patients that are more different) in order to achieve balance across the trial and control groups. The advantage to this process is that it retains all of the patients in both groups, however, the downside is that patients now represent fractions (larger or smaller) of their original selves, which is more difficult to explain to a broad stakeholder audience. On the AEP, we have the flexibility to choose between multiple adjustment methods, which allows us to implement the ideal method for each individual ECA or study. Below are figures that represent a fictional original, unbalanced population, and the impact of both adjustment methods.
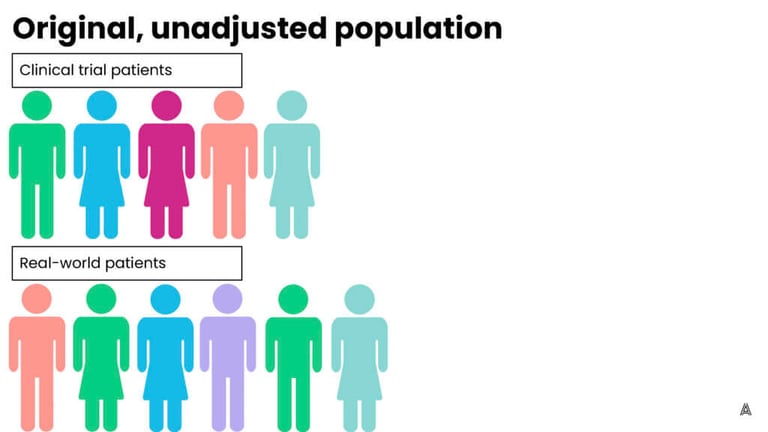


Step 5: Assess the results
Lastly, we assess the results to identify key differences in outcomes between the clinical trial and the ECA, and draw conclusions about the benefits (or lack thereof) of the intervention in question compared to standard of care.
Drive impact for future drug approvals
ECAs have the potential to completely transform the way that we approve drugs and assess their effectiveness, and possibly even shorten the time for a critical treatment to come to market. In addition, ECA studies provide comparative results that could never be discerned from a single-arm trial, which allows regulators, clinicians, and patients to make more informed decisions around treatment choice. However, it is crucial to recognize the complexities of these studies, and the challenges to constructing a methodologically sound ECA study. It requires input from experts across data strategy, data science, biostatistics, and advanced pharmacoepidemiology groups, and a platform or program to actually execute the ECA.
Here at Aetion, we are proud to have supported our clients in creating ECAs across multiple disease areas, using the AEP. We look forward to continuing to work on ECAs as they emerge as standard practice to support biopharma, regulators, and ultimately patients and clinicians in making better informed decisions.